Contenidos
- Cancer sintomas .com
- Cáncer de piel
- Cáncer de tiroides
- Cáncer de colon
- Cáncer de mama
- Cáncer de garganta (laringe)
- Cáncer de estómago
- Cáncer de vagina (vaginal)
- Cáncer oral (de boca)
- Cáncer de próstata
- Cáncer de huesos (óseo)
- Cáncer de vulva (vulvar)
- Cáncer de pulmón
- Cáncer de útero
- Cáncer de vejiga
- Cáncer de hígado
- Cáncer de ano (anal)
- Cáncer de pene
- Leucemia (cáncer en la sangre)
- Tumor cerebral (cáncer de cerebro)
- Cáncer de páncreas
- Cáncer rectal
- Mieloma múltiple
- Cáncer de riñón
- Cáncer de vesícula biliar
- Cáncer de glándulas salivales
- Cáncer de cuello uterino (cervical)
- Cáncer de ovario
- Cáncer de ojo (ocular)
- Cáncer de esófago (esofágico)
- Cáncer de corazón
- Cáncer de testículo
- Linfoma (de Hodgkin y no)
- Retinoblastoma
- Tumor de Wilms
- Neuroblastoma
- Mesotelioma
- Rabdomiosarcoma
- Imágenes contra el cáncer
- Asociación Americana del Cáncer (Sociedad ACS)
Versão em português
También de interés
Publicidad
Síntomas de neuroblastoma
Signos de neuroblastoma, información sobre las señales de neuroblastoma infantil, qué es.
El neuroblastoma es un cáncer que comienza en las células nerviosas inmaduras del sistema nervioso simpático. Estas células son llamadas neuroblastos, o células de la cresta neural. El sistema nervioso simpático se encarga de preparar el cuerpo para entrar en acción y responder a situaciones donde se necesita fuerza y conciencia, como aquellas que causan miedo, ira, emociones o vergüenza. A veces, los neuroblastos cambian y producen tumores benignos como el ganglioneuroma. En algunos casos, los cambios en los neuroblastos pueden causar neuroblastomas.
Se puede desarrollar en cualquier parte del sistema nervioso simpático, pero comienza con mayor frecuencia en las glándulas suprarrenales, el cuello, el pecho o la médula espinal. El neuroblastoma casi siempre se desarrolla en los bebés y niños pequeños.
Un neuroblastoma puede ocurrir en cualquier parte a lo largo del sistema nervioso periférico, por lo que los signos y síntomas de la enfermedad varían con la ubicación. Los síntomas más comunes son causados por la presión del tumor en los tejidos cercanos a medida que crece, o por la propagación del cáncer a los huesos. En la mayoría de los casos, el neuroblastoma no causa problemas en el funcionamiento del sistema nervioso periférico.
Los niños con neuroblastoma abdominal podrían tener los siguientes signos y síntomas: abdomen hinchado, estreñimiento, dificultad para orinar, aumento de la presión arterial, náuseas y vómitos, pérdida de apetito, pérdida de peso, quejas de plenitud en el abdomen (estómago hinchado), e hinchazón de la ingle o las piernas.
Los niños con neuroblastoma en el pecho podrían tener los siguientes síntomas: dificultad para respirar o falta de aliento, dificultad para tragar y tos.
Los niños con neuroblastoma en el cuello podrían tener los siguientes síntomas: bulto visible en el cuello, dificultad para respirar, dificultad para tragar, una mezcla de colores en el iris de los ojos (heterocromía) y síndrome de Horner (raro). El síndrome de Horner se caracteriza por párpados caídos (ptosis ipsilateral), pupilas pequeñas (miosis), e imposibilidad de sudar en el lado afectado de la cara (anhidrosis) debido a daños en el nervio simpático del ojo de ese lado de la cara.
Los niños con neuroblastoma en la médula espinal podrían tener los siguientes síntomas: debilidad en las piernas, inestabilidad al caminar, incontinencia o dificultad para orinar o defecar, dolor y parálisis.
 |
Las células del neuroblastoma son poco diferenciadas (no se parecen a las células normales) y altamente malignas. Sin embargo, el neuroblastoma es uno de los pocos cánceres que a veces puede remitir espontáneamente. Esto significa que el tumor se hace más pequeño o desaparece completamente sin ningún tratamiento y sin que se desarrollen nuevos tumores. Se presenta con mayor frecuencia en niños menores de un año.
Los sitios más comunes donde puede extenderse el neuroblastoma son: los ganglios linfáticos, la médula ósea, los huesos (el neuroblastoma es conocido por su propagación a los huesos de la órbita del ojo, llamados huesos periorbitales), el hígado, y más raramente la piel (se caracteriza por un número variable de bultos azulados debajo de la piel), el cerebro y los pulmones.
La compresión de la médula espinal es un problema grave que puede ocurrir debido a un neuroblastoma. Los neuroblastomas que se desarrollan en el tejido nervioso a lo largo de la médula espinal pueden presionar o crecer sobre la médula espinal, causando dolor, incontinencia, debilidad en las extremidades y parálisis. Es importante diagnosticar y tratar el neuroblastoma lo antes posible porque la compresión de la médula espinal podría conducir a una parálisis permanente.
Ganglioneuromas
Un ganglioneuroma es un tumor benigno cuyas células son similares a las de las células de los neuroblastomas malignos, pero son más maduras (bien diferenciadas). Los ganglioneuromas suelen ser inofensivos y no causan ningún problema ni necesitan ningún tratamiento a menos que estén afectando a órganos o nervios cercanos. A veces, un neuroblastoma canceroso se convierte en un ganglioneuroma por sí solo o con tratamiento. No se sabe la causa de los ganglioneuromas ni qué factores de riesgo aumentan las probabilidades de que un niño lo desarrolle. Los signos y síntomas de un ganglioneuroma pueden ser similares a los de un neuroblastoma y dependerán de la ubicación del tumor.
Ganglioneuroblastoma
 |
FACTORES DE RIESGO PARA EL NEUROBLASTOMA
El neuroblastoma es el tumor sólido más común fuera del cerebro en los niños. Representa del 8% al 10% de todos los cánceres en niños. Alrededor del 80% de los neuroblastomas se producen antes de los 5 años, y la edad media de diagnóstico es de 2,5 años. Es el cáncer más común en los bebés menores de 1 año. El neuroblastoma es muy poco frecuente en niños mayores de 10 años de edad. Este cáncer presenta una frecuencia ligeramente mayor en niños que en niñas.
Hay pruebas convincentes de que los siguientes factores incrementan el riesgo de neuroblastoma:
Antecedentes familiares
Aproximadamente el 1%-2% de los niños diagnosticados con neuroblastoma tienen antecedentes familiares de la enfermedad. El riesgo de neuroblastoma parece ser más alto para los hermanos o gemelos idénticos que ya tienen la enfermedad.
Trastornos genéticos
Los trastornos genéticos son causados por cambios en ciertos cromosomas. Estos cambios en los cromosomas se transmiten de padres a hijos. El neuroblastoma se desarrolla en niños con trastornos genéticos que implican cambios en las células nerviosas inmaduras (células de la cresta neural, o neuroblastos):
- Enfermedad de Hirschsprung. Es un trastorno en el que los nervios están ausentes en parte de los intestinos. Como resultado, el intestino grueso no funciona adecuadamente y puede bloquearse.
- Síndrome de hipoventilación central congénita (también llamado hipoventilación alveolar primaria). Es un trastorno raro que afecta a la respiración. Las personas con este trastorno tienen respiraciones superficiales, especialmente cuando están durmiendo.
- Neurofibromatosis tipo 1 (enfermedad de von Recklinghausen). Es un trastorno en el que se desarrollan tumores no cancerosos en los nervios y la piel, causando que áreas de la piel se vuelvan más claras u oscuras.
- Síndrome de Beckwith-Wiedemann. Afecta al crecimiento de diferentes partes del cuerpo. Causa gran tamaño corporal, lengua grande, órganos grandes, un defecto en la pared abdominal y bajo nivel de azúcar en la sangre de los recién nacidos.
- Síndrome DiGeorge. Provoca un escaso desarrollo de varios sistemas del cuerpo, lo que lleva a problemas médicos como defectos cardíacos, paladar hendido y problemas emocionales y de comportamiento.
Hay otros factores que posiblemente pueden provocar neuroblastoma:
Consumo de alcohol durante el embarazo
Tomar alcohol durante el embarazo interrumpe el desarrollo normal de las células nerviosas inmaduras llamadas neuroblastos o células de la cresta neural. Algunos estudios han demostrado un vínculo entre el neuroblastoma y el consumo de alcohol antes o durante el embarazo.
Uso de medicamentos durante el embarazo
Los investigadores han analizado el uso de medicamentos durante el embarazo, incluyendo diuréticos, medicamentos para el dolor, medicamentos anticonvulsivos, anticonceptivos orales y hormonas de la fertilidad. Los resultados varían, pero son lo suficientemente fuertes como para mantener una posible relación entre algunas medicinas y el neuroblastoma.
Trabajo del padre
Los estudios han demostrado una posible relación entre la ocupación del padre y el neuroblastoma. Los niños cuyos padres trabajan en puestos de trabajo relacionados con la electrónica o en empleos que los exponen a campos electromagnéticos, hidrocarburos o polvo de madera, pueden tener más riesgo de neuroblastoma.
Factores desconocidos
Hay otros factores cuyo efecto aún no se conoce bien si pueden provocar neuroblastoma:
- exposición a virus o factores ambientales (como los productos químicos o radiación) durante el embarazo o después del nacimiento;
- trabajo de la madre en ciertas industrias (incluyendo agricultura, floristería, jardinería, peluquería).
Cuando el neuroblastoma se encuentra y se trata a tiempo, las posibilidades de éxito del tratamiento son mejores. Lleve a su hijo a controles regulares de salud y consulte con el médico si el niño tiene dificultad para respirar o falta de aliento, un bulto o hinchazón en el cuello o el abdomen, pérdida de control del intestino o la vejiga, dolor en el abdomen, o inestabilidad al caminar.
Los estudios realizados en Canadá, Europa y Japón no respaldan la detección del neuroblastoma mediante pruebas de orina. Los ensayos clínicos han demostrado que la detección en niños a una edad temprana (3 semanas, 6 meses y 1 año) no fue beneficiosa. Sólo se encontraron tumores que normalmente desaparecieron por su cuenta (regresión espontánea) o tumores que necesitaban muy poco tratamiento.
CÓMO SE DETECTA EL NEUROBLASTOMA
Hay diferentes métodos y pruebas para conseguir la detección de un neuroblastoma (diagnóstico):
- Examen físico. El médico puede: palpar el abdomen en busca de un bulto o agrandamiento del hígado, palpar ganglios linfáticos inflamados, examinar la piel en busca de bultos azulados debajo de la piel que se mueven, observar si el niño tiene ojos saltones (proptosis) y hematomas alrededor de los ojos (equimosis periorbital), y comprobar los brazos y las piernas del niño para ver si existe debilidad y/o entumecimiento.
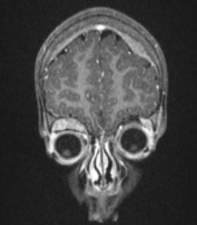 en niño de 2 años |
- Análisis de sangre completo. Un conteo sanguíneo completo se realiza para comprobar bajos niveles de glóbulos rojos, glóbulos blancos y plaquetas, lo que puede ser un signo de crecimiento del neuroblastoma en la médula ósea. Además, se hace una prueba química para detectar el aumento de los niveles de dopamina y noradrenalina, que puede ser un signo de neuroblastoma.
- Exámenes de sangre para los niveles de ferritina. El cuerpo almacena hierro en forma de ferritina. Como parte de la hemoglobina (una proteína de las células rojas de la sangre), el hierro se utiliza para transportar oxígeno en la sangre. Los niveles de ferritina se comprueban para diagnosticar el neuroblastoma y determinar un pronóstico.
- Pruebas de marcadores tumorales. Los marcadores tumorales son sustancias (generalmente proteínas derramadas en la sangre por las células tumorales) que pueden indicar la presencia de neuroblastoma. Estas pruebas se pueden utilizar para comprobar la respuesta de un niño al tratamiento del cáncer, pero también se pueden usar para ayudar a diagnosticar el neuroblastoma. Los marcadores tumorales que pueden medirse son: enolasa específica de la neurona (una enzima producida por las células neuronales y los tumores de neuroblastoma), GD2 (un gangliósido de la membrana celular que se encuentra en la superficie de la célula de neuroblastoma y parece acelerar el crecimiento del tumor), cromagranina A (una proteína producida por las células de neuroblastoma) y lactato deshidrogenasa (una enzima que puede ser producida por las células del neuroblastoma).
- Rayos X. Una radiografía sirve para encontrar el tumor en el cuerpo del niño y ayudar a decidir si el cáncer se ha propagado a los huesos.
- Ultrasonido. Observar las imágenes de las estructuras internas del cuerpo ayuda a encontrar el neuroblastoma en el cuerpo del niño.
- Tomografía computarizada (TC). Se utiliza para ayudar a localizar la posición y el tamaño del neuroblastoma, decidir si el tumor se puede extirpar mediante cirugía, y averiguar la extensión de la enfermedad y si se ha diseminado a otros órganos y tejidos. Las tomografías computarizadas pueden realizarse en el pecho, el abdomen, la pelvis, la cabeza, el cuello, la columna vertebral, o en cualquier área donde el médico sospecha que el cáncer se ha diseminado a los ganglios linfáticos.
- Resonancia magnética. Se utiliza para averiguar el tamaño del tumor y decidir si se puede extirpar mediante cirugía, ver si el cáncer se ha diseminado a otras partes del cuerpo y si los tumores cerca de la médula espinal están presionando sobre los nervios.
- Biopsia. Las biopsias que podrían ser utilizadas para el neuroblastoma son: biopsia con aguja gruesa (eliminación de tejido del neuroblastoma usando una aguja, biopsia incisional (extirpación quirúrgica de parte del tumor) y biopsia por escisión (extirpación quirúrgica de todo el tumor).
- Aspiración de médula ósea y biopsia. Consiste en tomar una muestra de la médula ósea (que se encuentra en el centro de los huesos y es donde se forman las células sanguíneas). Generalmente las muestras se toman de ambas caderas. Una biopsia de médula ósea puede mostrar enfermedad en la médula y confirmará si el neuroblastoma se ha diseminado desde el tumor principal.
- Gammagrafía ósea. Utiliza materiales radiactivos para el hueso (radiofármacos) y una computadora para crear una imagen de los huesos. El tecnecio 99 es el radiofármaco que se puede utilizar en una gammagrafía ósea para averiguar si el neuroblastoma se ha extendido a los huesos.
- Exploración MIBG. Utiliza el radiofármaco metayodobencilguanidina (MIBG), que contiene pequeñas cantidades de yodo radioactivo, para encontrar el tumor principal del neuroblastoma. También se utiliza para determinar si la enfermedad se ha diseminado a otros tejidos o huesos. Además, las exploraciones MIBG ayudan a mostrar si el tumor está respondiendo al tratamiento. Aproximadamente el 10% de los niños con neuroblastoma tienen tumores que no son detectados por la exploración MIBG (son MIBG negativo).
- Examen y biopsia de los ganglios linfáticos. Se examinan los ganglios linfáticos inflamados y se toman biopsias de los mismos. Los ganglios linfáticos que no se pueden palpar suelen ser evaluados por tomografía computarizada para obtener una imagen en 3 dimensiones y decidir si se les hace o no una biopsia. La biopsia de ganglios linfáticos se puede hacer si un ganglio linfático inflamado es la única área posible donde se ha extendido el tumor o si sería difícil o arriesgado hacer una biopsia del tumor primario. Esta biopsia se puede hacer a través de aspiración con aguja fina, con aguja gruesa, por biopsia incisional o por biopsia de escisión para extirpar todo el ganglio linfático. Este es el tipo más común de biopsia que se usa para detectar ganglios linfáticos para el neuroblastoma y, por lo general, se realiza en el momento de la cirugía.
TRATAMIENTO DEL NEUROBLASTOMA
Las decisiones de tratamiento para el neuroblastoma se basan en categorías de riesgo. Los niños con neuroblastoma se dividen en grupos de riesgo bajo, intermedio y alto. Estos grupos de riesgo se determinan según la edad del niño, la etapa del cáncer, y las características de las células del neuroblastoma. Estas características de las células pueden ser:
- copias extra del gen MYCN (amplificación MYCN);
- un aspecto favorable o desfavorable bajo el microscopio (histología);
- una cantidad normal o anormal de ADN (ploidía):
- una parte perdida del cromosoma 1p o 11q (en algunos casos).
Las opciones de tratamiento para el neuroblastoma son:
Cirugía
La cirugía se utiliza para establecer el diagnóstico, obtener tejido tumoral para su examen, conocer la etapa de la enfermedad y, si es posible, extirpar el neuroblastoma por completo. Una cirugía de segunda exploración puede hacerse después de varios ciclos de quimioterapia para eliminar el neuroblastoma restante y evaluar la forma en que el tumor está respondiendo al tratamiento.
Quimioterapia
 usadas para probar tratamiento |
Terapia de radiación
La radioterapia se puede administrar por sí sola o en combinación con quimioterapia, antes o después de la cirugía, para controlar un neuroblastoma que no se ha diseminado a otras partes del cuerpo. También se puede usar después de la cirugía para tratar el área donde ha sido retirado un neuroblastoma primario. La radiación puede usarse para reducir rápidamente los neuroblastomas en el hígado o en la columna vertebral si están causando síntomas que amenazan la vida. En casos raros, la radiación también puede utilizarse para tratar de reducir los neuroblastomas en torno a la cuenca del ojo, si el paciente ha quedado ciego o tiene problemas de visión significativos. También puede utilizarse para tratar áreas de metástasis óseas en niños con neuroblastoma de alto riesgo. La radioterapia paliativa se usa para aliviar los síntomas del cáncer avanzado.
Trasplante de células madre
A los niños que aún están en alto riesgo de neuroblastoma recurrente después de haber sido tratados con quimioterapia convencional y radioterapia, se les puede hacer un trasplante de células madre.
Terapia biológica
La terapia biológica se utiliza a menudo como terapia de mantenimiento al final del tratamiento, en la que se usan principalmente una clase de fármacos conocidos como retinoides.
La inmunoterapia es un tipo de terapia biológica usada para estimular la respuesta inmune del cuerpo y rechazar y destruir el neuroblastoma. La inmunoterapia puede ser utilizada como parte de la terapia de mantenimiento en niños con neuroblastoma de alto riesgo después de un trasplante de células madre.
Espera vigilante
En ciertas situaciones de bajo riesgo, la conducta expectante puede ser el tratamiento de elección. El seguimiento después de finalizar el tratamiento es importante, con visitas regulares de seguimiento, especialmente en los primeros 2 años después del tratamiento.
ESPERANZA DE VIDA Y SUPERVIVENCIA PARA EL NEUROBLASTOMA
La supervivencia a 5 años observada para el neuroblastoma en niños de 0-14 años de edad es del 77%. Esto significa que, en promedio, el 77% de los niños diagnosticados con neuroblastoma siguen vivos 5 años después de su diagnóstico. Sin embargo, la supervivencia varía según las categorías de riesgo de neuroblastoma (bajo un 95%, intermedio un 85-90% y alto un 30%). En general, cuanto antes se diagnostique y se trate el neuroblastoma, y más joven sea el niño, mejor será el resultado. Sin embargo, los neuroblastomas no suelen encontrarse hasta que están en una etapa avanzada, o tienen características desfavorables que pueden hacer que sean más difíciles de tratar.