Contenidos
- Cancer sintomas .com
- Cáncer de piel
- Cáncer de tiroides
- Cáncer de colon
- Cáncer de mama
- Cáncer de garganta (laringe)
- Cáncer de estómago
- Cáncer de vagina (vaginal)
- Cáncer oral (de boca)
- Cáncer de próstata
- Cáncer de huesos (óseo)
- Cáncer de vulva (vulvar)
- Cáncer de pulmón
- Cáncer de útero
- Cáncer de vejiga
- Cáncer de hígado
- Cáncer de ano (anal)
- Cáncer de pene
- Leucemia (cáncer en la sangre)
- Tumor cerebral (cáncer de cerebro)
- Cáncer de páncreas
- Cáncer rectal
- Mieloma múltiple
- Cáncer de riñón
- Cáncer de vesícula biliar
- Cáncer de glándulas salivales
- Cáncer de cuello uterino (cervical)
- Cáncer de ovario
- Cáncer de ojo (ocular)
- Cáncer de esófago (esofágico)
- Cáncer de corazón
- Cáncer de testículo
- Linfoma (de Hodgkin y no)
- Retinoblastoma
- Tumor de Wilms
- Neuroblastoma
- Mesotelioma
- Rabdomiosarcoma
- Imágenes contra el cáncer
- Asociación Americana del Cáncer (Sociedad ACS)
Versão em português
También de interés
Publicidad
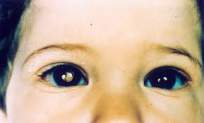
Síntomas de retinoblastoma
Signos de retinoblastoma, información sobre las señales de retinoblastoma infantil, qué es.
El retinoblastoma es un tumor maligno que comienza en las células de la retina del ojo.
Los signos y síntomas del retinoblastoma incluyen:
- Reflejo blanco o resplandor en una o ambas pupilas del niño cuando se exponen a la luz (como en una fotografía tomada con un flash). Toda la pupila suele aparecer blanca o rosa. El resplandor blanco de la pupila se llama leucocoria.
- Estrabismo (los ojos no parecen mirar en la misma dirección) o movimientos de los ojos no coordinados.
- Enrojecimiento e inflamación de la parte blanca del ojo.
- Visión borrosa. Los niños pueden toparse con cosas porque no pueden ver con claridad. A veces no pueden comer alimentos en un lado de su plato o jugar con los juguetes porque parte de su visión está bloqueada.
- Dolor en los ojos.
- Bizquera.
- Agrandamiento del ojo (buftalmos).
- Ojo que sobresale de la órbita del ojo (proptosis).
 |
- Heterocromía. Cada iris es de un color diferente.
- Hifema. Sangre delante del iris.
- La pupila no disminuye (no se estrecha) cuando se expone a la luz brillante.
- Dolores de cabeza. Pueden estar asociados con retinoblastoma trilateral (un tipo de cáncer cerebral infantil que puede desarrollarse junto con al retinoblastoma en ambos ojos).
¿Qué es el retinoblastoma?
La retina es la parte del ojo que le permite ver, una capa interna de células en la parte posterior del globo ocular. Se compone de las células nerviosas especiales que son sensibles a la luz. Cuando los ojos están en desarrollo tienen células llamadas retinoblastos. Estas células se dividen en nuevas células y llenan la parte del ojo que se convertirá en la retina. A veces hay un cambio o mutación en un gen de un retinoblasto llamado gen del retinoblastoma (gen RB1). Este gen mutado hace que el retinoblasto cambie y crezca fuera de control. Las células anormales causadas por esta mutación forman un tumor llamado retinoblastoma. El retinoblastoma es el tipo más común de cáncer de ojo en los niños. Se encuentra generalmente en los niños menores de 2 años.
Otros tipos de células en el ojo también pueden cambiar y provocar afecciones o tumores benignos que tienen algunos de los síntomas que el retinoblastoma, como las cataratas congénitas, el pseudoretinoblastoma, la retinopatía del prematuro y el retinocitoma.
TIPOS DE RETINOBLASTOMA
El retinoblastoma se puede clasificar en función de su causa, de cómo crece y de dónde se encuentra.
Retinoblastomas clasificados por causa
Tres tipos de retinoblastoma son clasificados de acuerdo con cómo se produjo la mutación del gen RB1:
- Retinoblastoma no hereditario. En algún momento después de la concepción, el gen del retinoblastoma (RB1) mutó por casualidad (mutación esporádica) en una célula del retinoblasto del ojo. La mutación genética hace que el retinoblasto se divida fuera de control y forme un tumor canceroso. Sólo las células tumorales del retinoblastoma tienen la mutación del gen RB1. Alrededor del 60% de los retinoblastomas no son hereditarios y se producen solo en un ojo (retinoblastoma unilateral).
- Retinoblastoma hereditario esporádico. Nadie más en la familia ha tenido retinoblastoma sino que la mutación del gen RB1 ocurrió en el óvulo o el esperma de los padres (mutación germinal) antes de la concepción (unión del óvulo y espermatozoide). Todas las células del niño contendrán la mutación del gen RB1. Alrededor del 30% de todos los retinoblastomas son de este tipo y se producen en ambos ojos (retinoblastoma bilateral).
- Retinoblastoma familiar. Los niños con retinoblastoma familiar tienen un padre o familiar con retinoblastoma. El niño heredó la mutación y todas las células en el cuerpo contendrán una mutación del gen RB1 (mutación de la línea germinal). Alrededor del 10% de todos los retinoblastomas son retinoblastomas familiares y se producen en ambos ojos (retinoblastoma bilateral).
Retinoblastoma clasificado por cómo crece
Tres tipos de retinoblastoma son clasificados de acuerdo con cómo crecen:
- Retinoblastoma endofítico. Es un tumor que crece a través de la capa interna de la retina. Un patrón de crecimiento endófito se asocia generalmente con trozos de tumor que flotan en el humor vítreo y acuoso (siembra vítrea).
- Retinoblastoma exofítico. Es un tumor que crece entre las capas de la retina. A menudo causa un desgarro en la retina (desprendimiento de retina).
- Retinoblastoma difuso infiltrante. Es un tipo raro de retinoblastoma que no suele formar un tumor sino que las células tumorales crecen a lo largo de la retina. Este tipo de retinoblastoma tiende a ser de crecimiento lento y se encuentra generalmente en niños en edad escolar.
Retinoblastoma clasificado por dónde se produce
 |
- Retinoblastoma intraocular. El cáncer se encuentra sólo en el ojo. El retinoblastoma no se ha propagado a tejidos alrededor del ojo o a otras partes del cuerpo.
- Retinoblastoma extraocular. El cáncer se ha diseminado fuera del ojo a tejidos alrededor del ojo o a otras partes del cuerpo.
- Retinoblastoma trilateral. Se produce cuando un tumor en la glándula pineal (pineoblastoma) del cerebro se asocia con un retinoblastoma bilateral. La glándula pineal es una glándula en el cerebro que controla el ciclo de sueño y juega un papel en la maduración sexual. El retinoblastoma trilateral usualmente ocurre 20 meses o más después de que se diagnostica el retinoblastoma en ambos ojos. Se desarrolla en aproximadamente el 5%-15% de los niños con retinoblastoma.
DETECCIÓN PRECOZ DEL RETINOBLASTOMA
Si su hijo presenta síntomas de retinoblastoma y/o tiene antecedentes personales o familiares de esta enfermedad, será necesario consultar al médico para realizar algunas de las siguientes pruebas:
- Examen de la vista. Los exámenes se realizan poco después del nacimiento y luego con una frecuencia de pocos meses hasta los 5 años.
- Prueba de sangre para detectar la mutación del gen RB1 si un miembro de la familia la tiene. Los resultados de la prueba de sangre ayudarán al médico a decidir con qué frecuencia se deben hacer los exámenes de la vista.
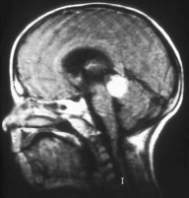
- Imágenes por resonancia magnética. A los niños diagnosticados con retinoblastoma hereditario se les hace una resonancia magnética para comprobar si hay retinoblastoma trilateral. El retinoblastoma trilateral puede desarrollarse unos 20 meses o más después del retinoblastoma. La resonancia magnética puede ser recomendada cada 6 meses durante los primeros 5 años de vida.
- Pruebas genéticas para RB1. Los niños y sus familiares que den positivo para la mutación en el gen RB1 pueden necesitar pruebas genéticas para determinar su riesgo de retinoblastoma. Esto es especialmente importante para los hermanos menores de 5 años del niño con retinoblastoma. Las personas con antecedentes personales o familiares de retinoblastoma que están planeando tener hijos también pueden desear hablar con su médico acerca de las pruebas genéticas y la evaluación del riesgo genético, que pueden ayudar a identificar una mutación en el gen RB1 que determinará si sus futuros hijos están en riesgo de desarrollar retinoblastoma. Las mujeres embarazadas también pueden optar por hacerse una prueba genética prenatal para el gen RB1, para ver si el feto tiene riesgo de desarrollar retinoblastoma.
CÓMO SE DETECTA EL RETINOBLASTOMA (DIAGNÓSTICO)
Además de revisar la historia clínica y los antecedentes familiares del niño, el médico puede hacer algunas de las siguientes pruebas:
Examen de la vista
Un examen ocular permite al médico revisar la visión, evaluar la salud de los ojos y buscar signos de retinoblastoma. A los niños se les suele dar anestesia o sedación antes de un examen completo de la vista para buscar un retinoblastoma.
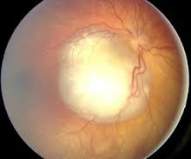 |
Se pueden poner gotas en los ojos para agrandar (dilatar) la pupila, lo que ayuda al médico a ver mejor las estructuras internas del ojo. Durante un examen ocular, el médico puede observar las diferentes estructuras del ojo para detectar anomalías (como sangrado), comprobar la parte anterior y parte posterior del ojo (el desprendimiento de retina es, a veces, una señal de retinoblastoma), buscar vasos sanguíneos inflamados en la superficie del ojo, verificar el movimiento del ojo y comprobar la presión dentro del ojo.
Pruebas genéticas
Las pruebas genéticas implican estudiar el ADN de una persona. Se realiza un análisis de sangre o tejido para buscar mutaciones de los genes del retinoblastoma 1 (RB1). Estas pruebas genéticas también se pueden hacer a otros miembros de la familia, como padres o hermanos del niño, sobre todo si hay antecedentes familiares de retinoblastoma o si se descubre que el niño tiene una mutación germinal. Una mutación germinal es una mutación del gen RB1 que sucedió en el óvulo o el esperma de los padres antes de la concepción (la unión del espermatozoide y el óvulo) y se transmite al niño.
Ultrasonido
El ultrasonido utiliza ondas sonoras de alta frecuencia para crear imágenes de estructuras internas del cuerpo. Un ultrasonido del ojo y de la órbita se utiliza para: confirmar un diagnóstico de retinoblastoma y averiguar la ubicación y el tamaño del tumor.
Una ecografía puede determinar el tamaño de retinoblastomas más grandes, pero no es tan útil para los tumores más pequeños. También se usa ultrasonido para ver hasta qué punto el tumor se extiende más allá del ojo y averiguar si hay más de un tumor en el ojo.
A veces se utilizan gotas para adormecer el ojo antes de realizar el ultrasonido. Los niños pueden recibir anestesia general o sedación antes del ultrasonido. La sonda de ultrasonido se coloca suavemente sobre los párpados cerrados o en la superficie del ojo.
Existen dos tipos de ultrasonidos que se pueden usar para examinar el ojo (ecografía oftálmica) y determinar el tamaño, características y alcance del retinoblastoma:
- Ecografía A. Proporciona una visión unidimensional del ojo. Se hace principalmente para medir la longitud del ojo.
- Ecografía B. Proporciona una vista en sección transversal, de dos dimensiones, del interior del ojo.
Resonancia magnética
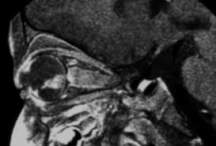 |
A veces se utiliza un medio de contraste con la resonancia magnética para ayudar a proporcionar un mejor detalle. Por lo general se inyecta en una vena de la mano o el brazo. Es importante que los niños no se muevan durante el procedimiento, por lo que se puede administrar anestesia general o sedación antes de la resonancia magnética.
Tomografía computarizada
Las tomografías se utilizan para determinar el tamaño del retinoblastoma (especialmente el retinoblastoma extraocular, es decir, el que se ha extendido más allá de los ojos), determinar el número de tumores, buscar depósitos de calcio en el ojo (calcificaciones oculares), y averiguar hasta qué punto el tumor se ha extendido más allá del ojo.
Un medio de contraste puede ayudar a proporcionar un mejor detalle. Por lo general se inyecta en una vena de la mano o el brazo. Es importante que los niños se queden quietos durante el procedimiento, por lo que se puede administrar anestesia general o sedación antes de la tomografía computarizada.
La tomografia no se usa tan a menudo como la resonancia debido a los niveles de radiación asociados con esta prueba.
Biopsia
A diferencia de la mayoría de otros tipos de cáncer, que necesitan una biopsia para confirmar el diagnóstico, el retinoblastoma generalmente se puede diagnosticar mediante un examen de los ojos y de imagen. La biopsia no es necesaria en muchos casos. Los médicos tienden a evitar tomar una biopsia del ojo, ya que puede ser difícil obtener una muestra del tumor sin dañar el ojo o sin propagar las células cancerosas dentro del ojo. Pero, en algunos casos, puede ser necesaria una biopsia si el retinoblastoma es muy difícil de diagnosticar.
La biopsia que se utiliza con mayor frecuencia es la aspiración con aguja fina. Una aguja y una jeringa muy delgada se utilizan para eliminar (aspirar) una pequeña cantidad de células del ojo. Se utilizan técnicas especiales para ayudar a prevenir la propagación del retinoblastoma si está presente.
Análisis sanguíneo completo
Se mide el número y la calidad de los glóbulos blancos, glóbulos rojos y plaquetas para ver si el retinoblastoma se ha diseminado fuera del ojo a la médula ósea.
Punción lumbar
Durante una punción lumbar, el médico inserta una aguja entre las vértebras lumbares inferiores de la columna para extraer una pequeña cantidad de líquido cefalorraquídeo y examinarla bajo un microscopio. Una punción lumbar se puede realizar para ver si el retinoblastoma se ha propagado fuera del ojo.
La punción lumbar no se hace muy a menudo durante el diagnóstico y estadificación del retinoblastoma. Se puede hacer si el nervio óptico o una gran parte de la coroides contiene cáncer, lo que sugiere que existe el riesgo de que el tumor se haya diseminado fuera del ojo.
Aspiración de médula ósea y biopsia
Durante una aspiración de médula ósea y biopsia, se extraen células de la médula ósea para que puedan ser examinadas en un laboratorio. El informe de anatomía patológica del laboratorio confirmará o no si el retinoblastoma se ha propagado a la médula ósea. Esta prueba se suele realizar únicamente si el nervio óptico contiene cáncer, lo que sugiere que el tumor se ha propagado fuera del ojo.
Gammagrafía ósea
La gammagrafía se utiliza para ver si el retinoblastoma se ha diseminado al cráneo o a otros huesos, aunque sólo se realiza si hay razones para creer que el cáncer se ha diseminado fuera del ojo. El niño puede recibir sedación antes de la gammagrafía ósea para que mantenga la calma y se esté quieto durante el procedimiento.
Angiografía con fluoresceína
La angiografía con fluoresceína es un tipo especial de procedimiento de rayos X utilizado para examinar los vasos sanguíneos en el interior del ojo. Se usa un tinte de color naranja especial llamado fluoresceína para hacer que los vasos sanguíneos del ojo sean visibles. El medio de contraste se inyecta en el brazo y viaja a los vasos sanguíneos del ojo. Luego se toman una serie de imágenes para ver cómo el cáncer está respondiendo al tratamiento (menos vasos sanguíneos suele significar que el cáncer está respondiendo al tratamiento) y para
predecir la evolución del niño después del tratamiento.
METÁSTASIS DEL RETINOBLASTOMA
El retinoblastoma generalmente se propaga por extensión directa del tumor o de la sangre. Los sitios más comunes hacia donde se extiende el retinoblastoma son:
- Otras partes del ojo, incluyendo la coroides, la conjuntiva, los párpados, la órbita (cavidad ocular) y el nervio óptico.
- Cerebro y médula espinal. Se propaga a través de las fibras nerviosas del nervio óptico directamente al cerebro. El cáncer a veces puede entrar en los espacios del cerebro que contienen el líquido cefalorraquídeo y, una vez allí, puede extenderse a cualquier parte del cerebro o de la médula espinal. El retinoblastoma también se puede propagar al cerebro a través de los vasos sanguíneos.
- Médula ósea, huesos largos del cuerpo, cráneo y senos paranasales.
- Ganglios linfáticos de la cabeza y el cuello.
- Pulmón e hígado.
TRATAMIENTO DEL RETINOBLASTOMA
Los objetivos del tratamiento del retinoblastoma (en orden de prioridad) son los siguientes: salvar la vida del niño, salvar los ojos y la visión del niño, y permitir que el niño vea bien después del tratamiento.
Las decisiones de tratamiento para el retinoblastoma se basan en: etapa del retinoblastoma, localización del tumor dentro del ojo, el número de tumores dentro del ojo, el tamaño del tumor o tumores dentro del ojo, si el cáncer se ha propagado fuera del ojo, si el cáncer se produce en un ojo (retinoblastoma unilateral) o en ambos ojos (retinoblastoma bilateral), y si el médico piensa que la visión del niño se puede salvar.
Los siguientes tratamientos se pueden ofrecer para el retinoblastoma:
Tratamientos focales
Los tratamientos focales pueden incluir criocirugía, termografía, cirugía láser o braquiterapia. Se utilizan en niños con retinoblastoma intraocular. También pueden ser utilizados para tratar un retinoblastoma que se repite dentro del ojo.
Quimioterapia
La quimioterapia puede administrarse como tratamiento primario, o combinada con tratamientos focales, para destruir las células cancerosas. Se puede administrar antes que otros tratamientos, como la criocirugía, la cirugía láser, la cirugía (extirpación del ojo o enucleación) o la radioterapia, para reducir el tamaño del tumor (quimioterapia neoadyuvante o quimiorreducción). Se puede administrar después de la cirugía, con radioterapia o sin ella, para destruir las células cancerosas remanentes y para reducir el riesgo de recurrencia de cáncer (quimioterapia adyuvante).
La quimioterapia se utiliza antes de un trasplante de células madre para tratar el retinoblastoma avanzado que se ha propagado fuera del ojo (retinoblastoma extraocular) o que es recurrente. También puede usarse para aliviar el dolor o para controlar los síntomas del retinoblastoma avanzado (quimioterapia paliativa).
Cirugía
La cirugía puede ser utilizada como tratamiento primario para curar el cáncer eliminándolo completamente. Se puede utilizar para eliminar la mayor cantidad de cáncer posible, sobre todo cuando la vista (visión) no se puede salvar. La cirugía puede utilizarse si otro tipo de tratamiento no funciona o si el cáncer regresa. También se puede utilizar si el cáncer conduce a glaucoma doloroso (aumento de la presión en el ojo) o a una pérdida de visión. Puede ser usada, finalmente, para colocar un catéter venoso central en los niños que pasarán por quimioterapia.
Radioterapia
La braquiterapia y la radioterapia de haz externo se pueden utilizar para tratar el retinoblastoma. La radioterapia se utiliza a veces como el principal tratamiento para destruir las células cancerosas. Se puede administrar después de la quimioterapia o de la cirugía para destruir las células cancerosas remanentes y reducir el riesgo de recurrencia del cáncer (radioterapia adyuvante). También se usa si otro tipo de tratamiento falla o el cáncer regresa, y para aliviar el dolor o para controlar los síntomas del retinoblastoma avanzado (radioterapia paliativa).
Trasplante de células madre
A los niños cuyo cáncer se ha propagado a sitios distantes en el cuerpo (retinoblastoma metastásico) se les puede dar un trasplante de células madre después de recibir altas dosis de quimioterapia. No se hace este transplante a menos que el retinoblastoma haya respondido a la quimioterapia. Se puede dar a los niños cuyo cáncer no ha respondido a otros tratamientos, como la cirugía y la radioterapia. Puede ser utilizada para tratar el cáncer que ha vuelto a aparecer (recurrente).
Seguimiento después de finalizar el tratamiento
Es importante hacer visitas regulares de seguimiento, especialmente en los primeros 3-5 años después del tratamiento.