Contenidos
- Cancer sintomas .com
- Cáncer de piel
- Cáncer de tiroides
- Cáncer de colon
- Cáncer de mama
- Cáncer de garganta (laringe)
- Cáncer de estómago
- Cáncer de vagina (vaginal)
- Cáncer oral (de boca)
- Cáncer de próstata
- Cáncer de huesos (óseo)
- Cáncer de vulva (vulvar)
- Cáncer de pulmón
- Cáncer de útero
- Cáncer de vejiga
- Cáncer de hígado
- Cáncer de ano (anal)
- Cáncer de pene
- Leucemia (cáncer en la sangre)
- Tumor cerebral (cáncer de cerebro)
- Cáncer de páncreas
- Cáncer rectal
- Mieloma múltiple
- Cáncer de riñón
- Cáncer de vesícula biliar
- Cáncer de glándulas salivales
- Cáncer de cuello uterino (cervical)
- Cáncer de ovario
- Cáncer de ojo (ocular)
- Cáncer de esófago (esofágico)
- Cáncer de corazón
- Cáncer de testículo
- Linfoma (de Hodgkin y no)
- Retinoblastoma
- Tumor de Wilms
- Neuroblastoma
- Mesotelioma
- Rabdomiosarcoma
- Imágenes contra el cáncer
- Asociación Americana del Cáncer (Sociedad ACS)
Versão em português
También de interés
Publicidad
Síntomas del tumor de Wilms
Signos iniciales y de tumor avanzado en niños (infantil), tratamiento y cura del tumor de Wilms, pronóstico, causas, complicaciones, mortalidad.
Los síntomas del tumor de Wilms son: un bulto indoloro o hinchazón en el abdomen, dolor en el abdomen, sangre en la orina (hematuria), hipertensión, fiebre, anemia, malestar estomacal, pérdida de peso, pérdida de apetito, venas del abdomen que parecen más grandes de lo normal, malestar general, debilidad, fatiga, y/o dificultad para respirar (si el cáncer se ha propagado a los pulmones).
El tumor de Wilms es un tumor canceroso del riñón que puede extenderse a otras partes del cuerpo. A veces no causa signos o síntomas en sus primeras etapas.
Los riñones están profundos en el cuerpo y no tienen muchos nervios, por lo que estos tumores pueden crecer sin causar dolor o malestar. Los síntomas aparecen una vez que el tumor crece en los tejidos y órganos. Otras trastornos de salud pueden causar los mismos síntomas que el tumor de Wilms.
QUÉ ES EL TUMOR DE WILMS
El tumor de Wilms es un tumor maligno que se origina en las células del riñón. Es el tipo más común de cáncer de riñón en niños.
El riñón es parte del sistema urinario. Tenemos dos riñones, uno a cada lado de la columna vertebral, muy dentro de la parte superior del abdomen. Los riñones producen orina filtrando el agua y los desechos de la sangre.
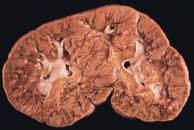 |
Los cambios en las células del riñón también pueden causar trastornos precancerosos. Esto significa que las células aún no son cáncer aunque hay una posibilidad más alta de que estos cambios anormales se conviertan en cáncer. El trastorno precanceroso más frecuente del riñón es la nefroblastomatosis. En la nefroblastomatosis hay muchos restos nefrogénicos. Un resto nefrogénico es un trozo de tejido embrionario del riñón que queda después de desarrollarse el embrión. Mientras que algunos restos nefrogénicos desaparecen por su propia cuenta, otros pueden convertirse en tumores de Wilms.
 |
En algunos casos, los cambios en las células de riñón pueden causar un tumor de Wilms. El tumor de Wilms se desarrolla a partir de los cambios en las células inmaduras (embrionarias) del riñón. Se encuentra generalmente en niños de 2 a 4 años.
También pueden desarrollarse otros tipos raros de cáncer de riñón en los niños como el sarcoma de células claras, el tumor rabdoide y el carcinoma de células renales.
Las células cancerosas pueden diseminarse desde el riñón a otras partes del cuerpo y se convierten en un nuevo tumor. Si el tumor de Wilms se propaga, es más probable que se extienda a los ganglios linfáticos cercanos a los riñones, la vena grande del abdomen que va al corazón (llamada vena cava inferior), los pulmones, el hígado u otro riñón.
TIPOS DE TUMORES DE RIÑÓN EN NIÑOS
Aunque el tumor de Wilms es el tipo de tumor renal maligno más común en niños, también pueden darse otros tipos más raros como el sarcoma de células claras del riñón, el tumor rabdoideo y el carcinoma de células renales.
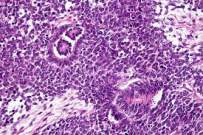
Tumor de Wilms
 |
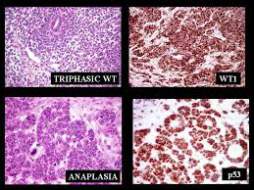 |
Alrededor del 90% de los tumores de Wilms se clasifican como de histología favorable. No hay anaplasia presente. Estos tumores con histología favorable responden mejor a la quimioterapia, por lo que tienen un mejor pronóstico que los que tienen una histología desfavorable.
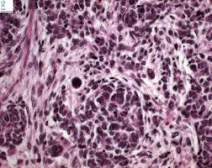 |
Sarcoma de células claras del riñón
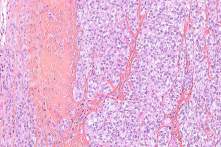 |
Carcinoma de células renales
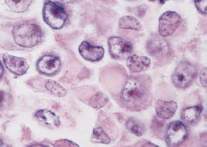
Tumor rabdoideo
 |
FACTORES DE RIESGO PARA EL TUMOR DE WILMS
Los expertos creen que el tumor de Wilms es causado por los cambios, o mutaciones, en ciertos genes. Un gen es la unidad básica de herencia que pasa de padres a hijos. Un trastorno genético es un conjunto de síntomas que ocurren juntos y, por lo general, son causados por uno o más genes anormales. Tener ciertos trastornos genéticos puede aumentar el riesgo de desarrollar un tumor de Wilms. Alrededor del 5%-10% de los niños diagnosticados con tumor de Wilms tienen un trastorno genético. El tumor de Wilms también es más común en niños con ciertos defectos congénitos o anomalías congénitas. Alrededor del 10% de los niños con tumor de Wilms tienen una anomalía congénita.
Hay pruebas convincentes de que los siguientes factores incrementan el riesgo para el tumor de Wilms:
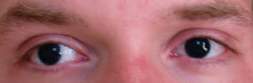
- Síndrome WAGR. Es un trastorno genético raro causado por la pérdida o inactivación de un gen supresor de tumor. Los genes supresores tumorales controlan el crecimiento celular y detienen el crecimiento de tumores. Cuando estos genes no están presentes o no funcionan correctamente, ya no controlan el crecimiento celular y se pueden formar tumores. Las siglas WAGR se refieren a los cuatro trastornos presentes en el síndrome: el tumor de Wilms, la Aniridia (cuando la parte coloreada del ojo, el iris, no se forma completamente), la presencia de malformaciones Genitourinarias (los sistemas urinario y reproductor no se forman normalmente) y Retraso mental. La mayoría de las personas que tienen el síndrome WAGR presentan dos o más de estos trastornos, pero no todas las personas con síndrome WAGR desarrollarán tumor de Wilms. Los niños con síndrome de WAGR tienen entre un 25% y un 30% de posibilidades de desarrollarlo.
- Síndrome de Beckwith-Wiedemann. Es un trastorno genético que causa que el cuerpo y algunos órganos, como la lengua, el hígado, y el bazo, crezcan más de lo normal. También afecta al vientre de los bebés, de manera que el abdomen no se cierra alrededor de la base del cordón umbilical. Como resultado, los órganos sobresalen en la base del cordón. Esto se llama onfalocele. Los niños con síndrome de Beckwith-Wiedemann tienen un riesgo del 5% de desarrollar un tumor de Wilms.
- Síndrome de Denys-Drash. Es un trastorno genético que causa enfermedad renal en el nacimiento y tumor de Wilms. También afecta a los órganos sexuales, que no se forman correctamente. Casi todos los niños con síndrome de Denys-Drash desarrollarán un tumor de Wilms en uno o ambos riñones. Por lo general se diagnostica alrededor de los 2 años de edad.
- Síndrome de Simpson-Golabi-Behmel. Es un trastorno genético que se encuentra sobre todo en los niños. Afecta a muchas partes del cuerpo. Los bebés con este síndrome son mucho más grandes de lo normal al nacer, y crecen y aumentan de peso a un ritmo inusual. Alrededor del 10% de los niños con síndrome de Simpson-Golabi-Behmel desarrollarán tumores benignos o cancerosos durante la infancia. El más común de estos tipos de cáncer es el tumor de Wilms.
- Síndrome de Perlman. Es una enfermedad genética muy rara y muy similar al síndrome de Beckwith-Weidemann. Se debe a que durante el embarazo hay demasiado líquido en el útero y, como resultado, el bebé puede nacer más grande y pesado de lo normal, con rasgos faciales anormales. Los bebés y los niños con síndrome de Perlman tienen un riesgo muy alto de padecer tumor de Wilms.
- Síndrome de Frasier. Es una enfermedad genética muy rara. En los niños, afecta a ambos riñones y causa que los genitales no se formen correctamente. En las niñas, por lo general, sólo afecta a los riñones.
 |
- Hemihipertrofia. Es un defecto congénito que causa que una parte o un lado del cuerpo crezca más grande que el otro.
- Testículos no descendidos (criptorquidia). Es un defecto de nacimiento común en los varones. Esto significa que uno o ambos testículos no descienden hacia abajo en el escroto.
- Hipospadias. Es un defecto de nacimiento en los niños. Se produce cuando la abertura urinaria (uretral) está en la parte inferior del pene en lugar de en la punta.
- Antecedentes familiares. Un pequeño número de los tumores de Wilms se desarrollan en niños que tienen familiares con la enfermedad. Aproximadamente el 1%-2% de los niños diagnosticados con tumor de Wilms tienen antecedentes familiares de la enfermedad.
- Pesar más de 4 kilos al nacer. Es un factor de riesgo para el tumor de Wilms, especialmente para las niñas. Un mayor peso al nacer se ha relacionado con el tumor de Wilms, pero no hay pruebas suficientes para demostrar que es un factor de riesgo conocido.
Se necesitan más estudios para ver si los siguientes son factores de riesgo para el tumor de Wilms: exposición de los padres a los pesticidas o a ciertos productos químicos industriales, y exposición de la madre a los tintes para el cabello o a ciertos medicamentos durante el embarazo o el parto.
DIAGNÓSTICO DEL TUMOR DE WILMS
El diagnóstico de tumor de Wilms generalmente comienza con una visita al médico de su hijo. El médico le preguntará acerca de cualquier síntoma que tenga su hijo y le realizará un examen físico. Con base en esta información, el médico puede derivarlo a un especialista u ordenar exámenes para verificar si existe tumor de Wilms u otros problemas de salud.
El proceso de diagnóstico puede parecer largo y frustrante. Es normal preocuparse, pero trate de recordar que otros trastornos de salud pueden causar síntomas similares a un tumor de Wilms. Es importante que el equipo de atención médica descarte otras causas de un problema de salud antes de hacer un diagnóstico de tumor de Wilms. Si el médico sospecha que es tumor de Wilms, el cuidado de su hijo estará a cargo de un equipo de salud que se especializa en el cuidado de niños con cáncer. Todos los casos de tumores renales infantiles se consideran urgentes.
Las siguientes pruebas se utilizan habitualmente para descartar o diagnosticar el tumor de Wilms:
- Antecedentes de salud y examen físico. Al tomar una historia clínica, el médico le hará preguntas sobre los antecedentes familiares de tumor de Wilms y la historia personal de su hijo con ciertos trastornos genéticos. El médico también le preguntará sobre la historia personal del niño de ciertas anomalías congénitas: aniridia, hemihipertrofia, testículos no descendidos, o hipospadias. El examen físico permite al médico detectar cualquier signo de tumor de Wilms. Durante el examen físico, el médico puede palpar el abdomen para notar cualquier bulto o hinchazón.
- Ultrasonido. Se utiliza para ver los riñones y las estructuras circundantes. Una ecografía abdominal es a menudo la primera prueba de imagen que se realiza si el médico sospecha que existe tumor de Wilms, ya que es un procedimiento indoloro y permite al médico ver todo el abdomen.
- Tomografía computarizada. Se utiliza para ver los riñones y también es útil para comprobar si el cáncer se ha diseminado a otros órganos o tejidos, como el otro riñón, los pulmones, el hígado o el cerebro.
- Resonancia magnética. Se utiliza para mostrar imágenes más detalladas de los riñones, incluyendo los principales vasos sanguíneos cerca del riñón. También se puede utilizar para comprobar si el cáncer se ha propagado al otro riñón o el cerebro.
- Pruebas de química sanguínea. Se pueden usar para diagnosticar la etapa del tumor de Wilms. Las pruebas de función renal muestran cómo están funcionando los riñones y las pruebas de función hepática hacen lo mismo con el hígado.
- Análisis de orina. Se puede hacer para ver si hay sangre en la orina y para comprobar los niveles de ciertas sustancias en la orina.
- Cirugía. En la mayoría de los casos de tumor de Wilms, lo mejor es eliminar el tumor primario sin hacer una biopsia. La cirugía más común es la nefrectomía, que elimina el tumor y el riñón. Esta cirugía permite a los médicos determinar la extensión de la enfermedad. El cirujano también tomará muestras de tejido de los ganglios linfáticos cercanos para determinar el estadio de la enfermedad y los mejores tratamientos después de la cirugía. Una biopsia del tumor de Wilms sólo se hará en lugar de la cirugía si el tumor no se puede quitar, el niño sólo tiene un riñón o hay tumores en ambos riñones. Las muestras recogidas durante la cirugía o la biopsia son examinadas en un laboratorio.
- Análisis de sangre completo. Se hace para verificar si hay anemia, que es un número bajo de glóbulos rojos.
- Radiografía. Una radiografía de tórax puede realizarse para comprobar si el cáncer se ha propagado a los pulmones.
TRATAMIENTO PARA EL TUMOR DE WILMS
El equipo de atención médica creará un plan de tratamiento específico para el niño con tumor de Wilms. Se basará en las necesidades del niño y puede incluir una combinación de diferentes tratamientos. Al decidir qué tratamientos ofrecen para el tumor de Wilms, el equipo médico tendrá en cuenta el tipo de tumor de Wilms (si se trata de histología favorable o desfavorable, la etapa del tumor, la edad del niño, la salud general del niño y los
cambios en los cromosomas.
Hay dos métodos principales para el tratamiento de niños con tumor de Wilms. Cada método tiene sus pros y sus contras.
En América del Norte, el enfoque estándar del Grupo de Oncología Infantil es extirpar el riñón afectado (nefrectomía) en el momento del diagnóstico, si es posible. Si el riñón no se puede extraer, se realiza una biopsia por lo general. Después de la cirugía para extirpar el riñón, o de la biopsia, los médicos examinarán las células tumorales para confirmar la histología y buscar cambios en los cromosomas. Se le da al tumor una etapa basada en las pruebas de imagen y en cómo las células tumorales se ven y actúan cuando se examinan bajo un microscopio. Se utiliza esta información para planificar el tratamiento. Entonces se dará quimioterapia, radioterapia o ambas basándose en la histología y el estadio del tumor.
El enfoque europeo es diagnosticar los tumores de riñón en base a estudios de imagen. La mayoría de los tumores renales se tratan con quimioterapia, y más tarde se extirpa el riñón afectado (por lo general después de 6 semanas de quimioterapia). El tratamiento adicional después de la cirugía para extirpar el riñón se basa en la etapa del tumor y en la forma en que ha respondido a la quimioterapia.
Las opciones de tratamiento para el tumor de Wilms son:
Cirugía
Es el tratamiento principal para el tumor de Wilms. Puede involucrar la nefrectomía radical o la nefrectomía parcial. La quimioterapia o la radioterapia pueden utilizarse junto con la cirugía.
Quimioterapia.
Se puede utilizar: después de la cirugía para prevenir la recurrencia (terapia adyuvante), antes de la cirugía (terapia neoadyuvante) para los tumores que no se pueden extirpar completamente, y para tratar tumores avanzados.
Los medicamentos de quimioterapia más comunes utilizados para tratar el tumor de Wilms son: vincristina,
dactinomicina, doxorrubicina, ciclofosfamida, cisplatino,
carboplatino y etopósido. Una combinación de estos medicamentos de quimioterapia es más eficaz que cualquiera de ellos por sí solo.
La ciclofosfamida puede irritar la vejiga. Cuando se utiliza este fármaco de quimioterapia, también se usa un medicamento llamado Mesna para proteger la vejiga.
Si el tumor de Wilms no responde a los medicamentos utilizados en los tratamientos anteriores o si se repite, hay otros regímenes de quimioterapia que se pueden administrar. Se usarán medicamentos en secuencia alterna o como quimioterapia de alta dosis, a veces con un trasplante de células madre.
Radioterapia
La radioterapia se administra en el abdomen tan pronto como sea posible, después de la cirugía, para extirpar el tumor. Los médicos se basan en la histología del tumor para determinar cuánta radiación se va a recibir. La radioterapia se administra 14 días después de la cirugía para extirpar el tumor de Wilms con histología favorable y 10 días después de la cirugía para extirpar el tumor de Wilms con histología desfavorable (anaplásico).
Las técnicas de radiación modernas permiten a los médicos apuntar a la zona a tratar con mucha más precisión, mientras se deja la mayor cantidad de tejido circundante normal posible. Estas técnicas son:
- Radioterapia conformacional tridimensional (3D-CRT). El oncólogo radioterapeuta utiliza imágenes de resonancia magnética para mapear la ubicación exacta y la forma del tumor. Se configuran varios haces de radiación y se dirigen al tumor desde diferentes direcciones para tratarlo desde todos los ángulos. Cada haz individual es bastante débil y menos propenso a dañar los tejidos normales. Una dosis más alta de radiación se dirige donde los rayos convergen en el tumor.
- Radioterapia de intensidad modulada (IMRT). Es similar a la 3D-CRT en que administra la radiación desde muchos ángulos diferentes para tratar el tumor entero. Además de dar forma y emitir los haces de radiación, esta técnica permite al oncólogo ajustar la fuerza o intensidad de los haces individuales. Esto hace que una dosis más alta llegue al tumor y se reduce la dosis de radiación que llega a los tejidos circundantes normales.
Seguimiento del tumor de Wilms
El seguimiento después de terminar el tratamiento permite que el equipo de salud vigile una posible recurrencia del tumor de Wilms y de los efectos secundarios tardíos del tratamiento. Los niños con tumor de Wilms deben tener visitas regulares de seguimiento, especialmente en los primeros 5 años después del tratamiento. Los niños que reciben radioterapia o algunos tipos de quimioterapia para el tumor de Wilms pueden necesitar un seguimiento durante toda la vida.
Es necesario informar al médico si su hijo tiene: un bulto o hinchazón en el abdomen, dolor en el abdomen, sangre en la orina, fiebre o pérdida de peso.
El seguimiento después de un tumor de Wilms suele ser programado: cada 3 meses durante 2 años después del diagnóstico, luego cada 6 meses durante otros 2 años, y luego una vez cada 2 años.
Las pruebas son a menudo parte de los cuidados de seguimiento: ecografía abdominal, tomografía computarizada, resonancia magnética, radiografía de tórax, análisis de sangre, pruebas de la función del corazón, ecocardiograma (eco) o electrocardiograma (ECG), análisis de sangre para verificar los niveles de la hormona estimulante de la tiroides (TSH) y la tiroxina libre (T4), etc.
Ensayos clínicos
Los ensayos clínicos permiten estudiar nuevas y mejores formas de prevenir, detectar y tratar el cáncer. El tumor de Wilms es poco frecuente, así que a muchos niños con este tipo de cáncer se les ofrecerá tratamiento en un ensayo clínico.
PRONÓSTICO Y SUPERVIVENCIA DEL TUMOR DE WILMS
La esperanza de vida a 5 años del tumor de Wilms en los niños de 0-14 años de edad es del 85%. Esto significa que, en promedio, el 85% de los niños con diagnóstico de tumor de Wilms se espera que sigan vivos 5 años después de su diagnóstico. No obstante, este porcentaje varía mucho según los siguientes son factores pronósticos y predictivos:
- Tipo de tumor. El tipo de tumor de Wilms es un factor pronóstico importante. Los tumores con histología favorable (sin anaplasia, lo que significa que las células se parecen y se dividen como las células normales) tienen un mejor pronóstico que aquellos con histología desfavorable (la anaplasia está presente). La histología anaplásico difusa (la anaplasia se encuentra en todo el tumor) se asocia con mayores tasas de recaída y una peor supervivencia que la histología favorable.
- Etapa. Cuanto más precoz sea la etapa del tumor de Wilms, más favorable será el pronóstico. El cáncer que se ha diseminado a los ganglios linfáticos o a otras áreas del cuerpo tiene un peor pronóstico.
- Edad. Una menor edad es un factor pronóstico más favorable. Los niños menores de 2 años de edad tienen un pronóstico significativamente mejor que los niños mayores. Esto puede ser debido al hecho de que la anaplasia no es común en niños menores de 2 años con diagnóstico de tumor de Wilms.
- Cambios en los cromosomas. Algunas células tumorales tienen ciertos cambios en los cromosomas que pueden afectar al pronóstico. La pérdida de heterocigosidad es cuando un alelo de un gen dentro de las células tumorales deja de funcionar. Esto puede significar que un gen que normalmente ayuda a limitar el crecimiento de las células cancerosas (llamado gen supresor de tumor) deja de funcionar. Los marcadores de heterocigosidad de ADN en los cromosomas 1p y 16q se han asociado con una mayor tasa de recurrencia y un peor pronóstico en pacientes con tumores de Wilms de histología favorable.
- Tamaño del tumor. Los tumores más pequeños tienen un pronóstico más favorable que los tumores más grandes.
- Síndromes genéticos. Varios síndromes genéticos, como el de Beckwith-Wiedemann, de Denys-Drash y WAGR, se asocian con el riesgo de desarrollar tumores de Wilms en ambos riñones. En general, los niños con estos síndromes genéticos tienen tumores que responden bien a la terapia y no aumenta el riesgo de recaída. Sin embargo, hay una incidencia mucho mayor de enfermedad grave del riñón (enfermedad renal terminal) en niños con tumores de Wilms en ambos riñones.
- Tumores de Wilms recurrentes. El pronóstico de un tumor de Wilms recurrente es generalmente mejor si el tumor tiene las siguientes características: histología favorable, en estadio I o II al momento del diagnóstico, sin quimioterapia previa con doxorrubicina, sin radioterapia anterior, y si la recurrencia se produce al menos 12 meses después del diagnóstico inicial.